| gene.not.interest | gene.in.interest | |
|---|---|---|
| In_category | 2613 | 28 |
| not_in_category | 15310 | 29 |
Enriquecimiento Funcional
La interpretación de una lista de genes diferencialmente expresados suele ser más util en terminos de vías biológicas.
- ¿Qué es una vía biológica? Una vía biológica es una serie de acciones entre las moléculas en una célula que genera un cierto producto o un cambio en la célula. Las células envían y reciben señales a traves de las vías biológicas.
Dado que los genes actuan colectivamente, un modelo más cercano a la biológia subyacente es aquel en el que las vías biológicas son las que se expresan diferencialmente.
Una aproximación sería intentar hacer el análisis de expresión diferencial directamente a nivel de vía biológica, o indirectamente buscando la sobrerepresentación de los genes diferencialmente expresados en cada vía de interes.
Bases de datos como Reactome o KEGG anotan información de la literatura sobre los genes y las reacciones que forman las vías biológicas. Otras bases de datos como The Gene Ontology (GO) y Molecular Signatures Database (MSigDB) definen las vías biológicas en terminos de conjuntos de genes.
Vías biológicas diferencialmente expresadas
Para encontras estas vía diferencialmente expresadas en terminos de conjunto de genes diferencialmente expresados, es detectanto un enriquecimiento funcional:
- Buscar genes diferencialmente expresados (DE).
- Para cada conjunto de genes, verificar si estos genes DE pertenecen a ese conjunto en una proporcion que excede la expectativa de encontrarlo solo por azar.
En el caso de que un conjunto de genes contiene más genes DE que los que se podrían esperar por puro azar, decimos que este conjunto de genes esta enriquecido en genes DE
Análisis de sobrerepresentación (ORA)
La forma de valorar si un conjunto de genes esta enriquecido o no es a traves de la prueba exacta de Fisher (prueba hipergeométrica). La prueba es útil para los datos categóricos que resultan de clasificar los objetos en dos formas diferentes. Se utiliza para examinar la significación de la asociación entre los dos tipos de clasificación. En nuestro caso si un gen es DE o no dentro de un conjunto de genes.
El valor de p se calcula con la función de distribución hipergeométrica. \[ \textrm{Pr}(X >= k)=\sum_{x=k}^{\textrm{min}(m, n)} \frac{ {m\choose x} {N-m\choose n-x} }{ {N\choose n} }\,. \]
- N: Número total de genes (background).
- m: Número de genes en la distribución que estan anotados en el conjunto de genes de interes.
- n: Tamaño de la lista de genes de interes.
- k: Número de genes de la lista que estan anotados en el conjunto de genes.
Ejemplo: Supongamos que tenemos 17980 genes detectados en un estudio de RNA-seq, y 57 genes estan diferencialmente expresados. Entre estos 28 estan anotados en un conjunto de genes.
Fisher's Exact Test for Count Data
data: df
p-value = 1
alternative hypothesis: true odds ratio is greater than 1
95 percent confidence interval:
0.110242 Inf
sample estimates:
odds ratio
0.1767937 Análisis de Gene Ontology
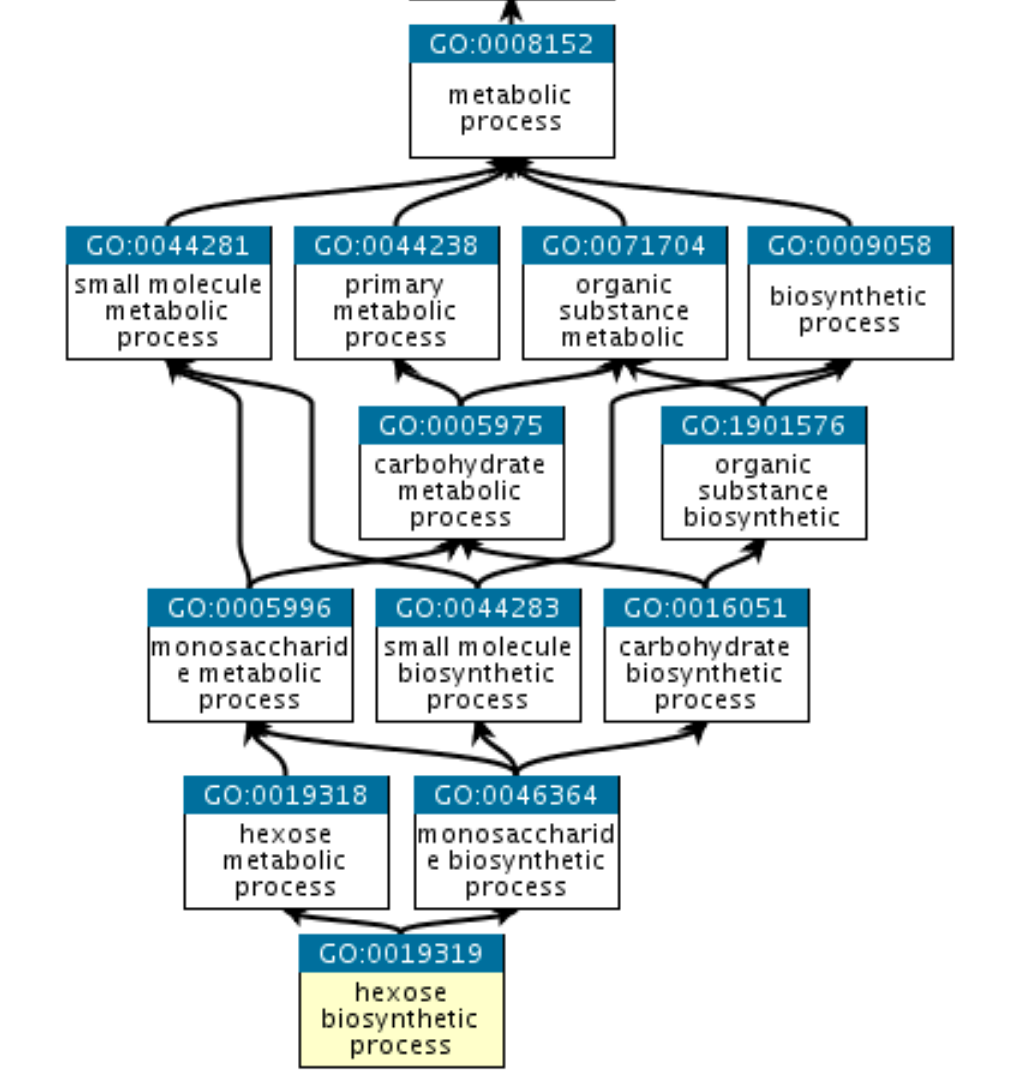
Es un análisis de enriquecimiento funcional aplicado a cada uno de los conjuntos de genes de la base da datos de GO Gene Ontology (GO) es una representación estructurada y estandarizada del conocimiento biológico. GO describe conceptos, tambien conocidos como términos, que estan interconectados a traves de relaciones definidas formalmente. GO esta diseñado para ser independiente de la especie.
Ontologías
Un término GO pertenece a una de las siguientes tres ontologías: Biological Process (BP), Molecular Function (MF) y Cellular Component (CC).
El producto de un gen puede asociarse con o localizarse en una o más componentes celulares y estar activo en uno o más procesos biológicos durante los cuales realiza una o más funciones moleculares.
Dentro de una ontología los términos GO estan relacionados unos con otros a través de relaciones jerarquicas que describen cuando un término GO es más general o nó.
Análisis de enriquecimiento de conjuntos de genes (GSEA)
El análisis de enriquecimiento ORA se basa en los genes identificados como diferencialmente expresados. Pero tiene la limitación de que necesita una lista minimamente grande de genes. Existen contextos donde los cambios de expresión diferencial puede ser pequeña. Los cambios moleculares más relevantes, si bien pequeños, ocurren de forma coordinada en una misma vía biológica. En estos contextos el método ORA suele fallar.
GSEA aborda directamente esta limitación. Todos los genes se pueden utilizar en GSEA (y no solamente los genes DE), esto hace posible detectar situaciones donde todos los genes en el conjunto predefinido cambian un poco pero de manera coordinada.
Descripción de GSEA
GSEA considera experimentos de perfiles de expresión de genes correspondientes a dos clases. Los genes son clasificados (genes L) basandose en la correlación entre su expresión y la distinción de clase usando una métrica adecuada (log2FoldChange o p-value).
Dado a priori un conjunto de genes S (p. ej., genes que comparten la misma categoría GO). El objetivo de GSEA es determinar si los miembros de los genes S estan aleatoriamente distribuidos a traves de los genes L o se encuentran principalmente en la parte alta o baja.
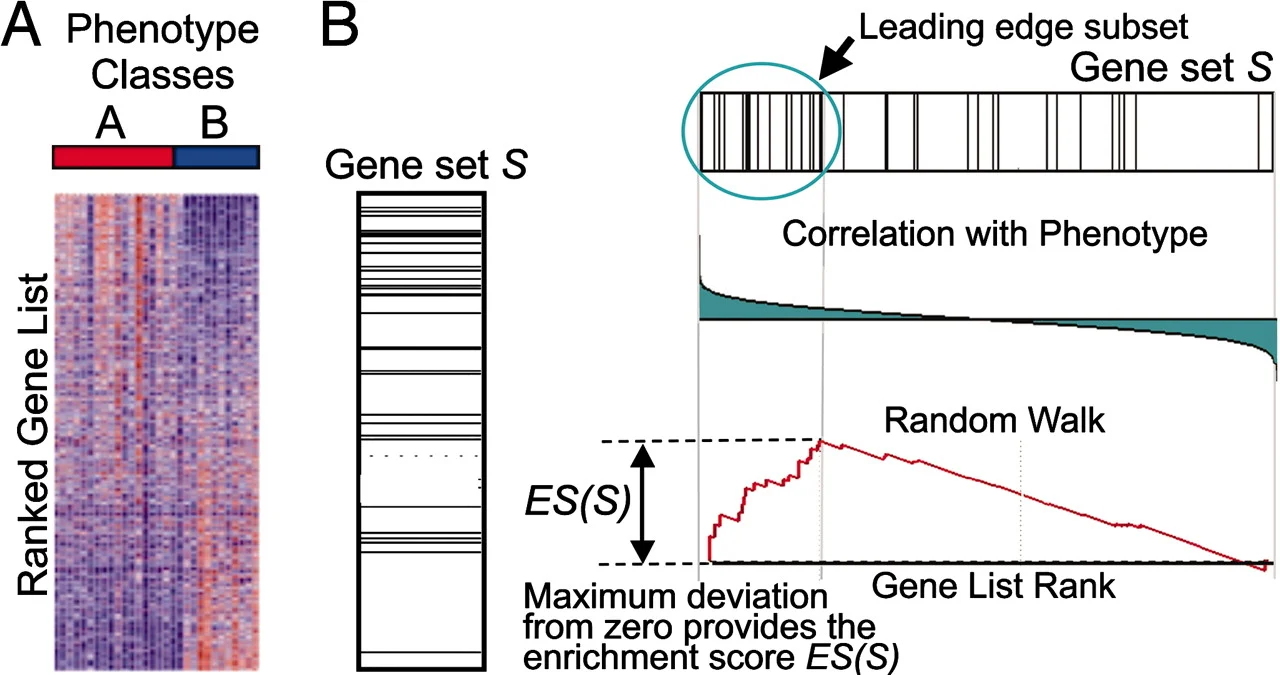
Hay 3 elementos clave en el metodo de GSEA:
Cálculo del Enrichment Score (ES): Se calcula el ES que refleja el grado por el cual un conjunto de genes S esta sobrerepresentado en los extremos de toda la lista de genes L. La magnitud del incremento depende de la correlación de los genes con el fenotipo.
Estimación del nivel de significancia de ES: Se calcula la significancía (p value) de ES usando un test de permutaciones basado en los fenotipos. Este test conserva la compleja estructura de correlación de los datos de expresión génica.
Ajuste de pruebas multiples: Se ajusta el nivel de significancia calculando el FDR para cada valor de NES (Normalized Enrichment Score).
Conjunto Leading-Edge
Los conjuntos de genes se pueden definir usando una variedad de métodos, pero no todos los miembros del conjunto de genes participan en los procesos biológicos. A veces es util extraer los genes que contribuyen al ES. Leading-edge, se define como el subconjunto de genes en los genes S que aparecen en la lista L antes, o despues del punto donde la suma alcanza la máxima desviación de cero.
References
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Enlace